|
|
|
Improvements and corrections in Mol2mol ver. 5.6.3:
(1.02.2011)
Chem3D Mullikan charges input - fixed
Problem with abbreviated structures input - fixed
Soft shadow (area lights) option added;
Black-and-white mode added, optionally with crosshatched atoms.
Dummy atom connectivity can be set to 0 now;
Dummy atom connectivity is taken into consideration when generating new connections.
Molden format (read);
Turbomole input format (read) and *.cosmo output (read);
Gamess and Q-chem input files are now recognized as normal files, so they can be copied and inputted via the clipboard, too.
The Help file and mol2mol.ini file have been reorganized. MDL V3000 RXN and RDF files sometimes failed to read - fixed.
Two rare but nasty bugs were eliminated when inputting multistructure CIFMIF or FDAT crystal files.
Sometimes the header of the output file was not correct because of an uninitialized variable - fixed;
In vector mode lone atoms without bonds are now drawn as spheres.
In some files the leading spaces of the lines caused problems - fixed;
Sometimes the coordinates in Angstrom or Bohr units were wrongly recognized - fixed;
Some third-party files were not recognized automatically - fixed;
Fine tuning of the input modul to accept some third party Gamess files.
Extra linefeeds were added to each line - fixed; Caption of an input dialog box was wrong - fixed.
Corrections in Mol2mol ver. 5.4.1:
(03.03.2005)
Display of H-bonds between the parent heavy atoms in molecules without hydrogens was not always correct - fixed
Improvements and corrections in Mol2mol ver. 5.4.0:
(20.02.2005)
Input of Gaussian output files (now recognized automatically);
Input of Gamess output files (now recognized automatically);
XMOL xyz format: Unichem subformat has been added (old but sometimes useful).
ZG-matrices sometimes crashed the program - fixed;
After clipboard input the conversion save file dialog did not open - fixed;
Nasty little hidden bug when removing dummy atoms - removed;
In PDB files if the 1st atom was HETATM the atom name was not always written - fixed.
Improvements and corrections in Mol2mol ver. 5.3.0:
(31.08.2004)
Calculation and dumping of named selections from HyperChem HIN or HCS files;
Calculation of the dihedral angle of two best fitted mean planes.
After using the menu option Open datafile | Current molfile to add partial charges the file was not closed - fixed;
Folder library: One-letter only nicknames were not accepted - fixed;
Clipboard output slowed down in the case much data - fixed;
Dumping of all dihedrals of an atom did not work in versions 5.1 - 5.2 - fixed.
Corrections and fixes in Mol2mol ver. 5.2.1:
(05.11.2003)
Improvements in Mol2mol ver. 5.2.0:
(15.10.2003)
bond types are supported in vector mode too;
support for stereo bond types;
support for rendering of elemental symbols and atom aliases in 2D mol files in vector mode. Example
Display of heteroatom symbols and atom aliases in vector models has been enhanced, a few small bugs fixed;
The SHELX edit mode could not be activated from the menu - fixed.
multiple pod-variables in the same line were skiped - fixed;
a few bugs in the calculation of bond colouring have been fixed;
Support of MacroModel atom types extended according to version 8.1, including generalized atom types;
In Macromodel multiple files sometimes the numbering and colouring of the models were not correct - fixed;
Not genuine PDB files: residue names were always written as UNK, atom names as elemental symbols. Now if the input file contains these, they are preserved and used.
Option to display hetero atom and hydrogen atom symbols in vector models;
Option added for choosing the anaglyph stereo colours: red/green or red/blue;
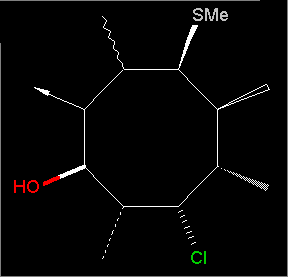
Stereo bond types are now displayed. Example
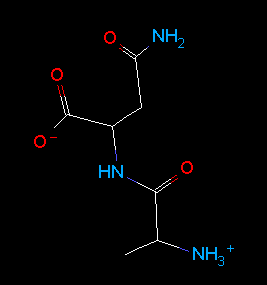
Implicit hydrogens are added and optionally shown; Example
Atom aliases from ISIS/Draw and ChemDraw ct files are displayed.
When converting to WinPLT files, implicit hydrogen support has been enhanced. Conversion of atom aliases is also supported.
Hydrogen addition in pH-dependent mode: -CONH2 group was also protonated - fixed
Z matrix writing: the program sometimes crashed with structures containing many small rings (complexes, crystalls) - fixed
Three/four-membered rings formed by special bond types were assigned to cyclopropyl/butyl - fixed
SDF/RDF files in browse mode: if the Data button was pressed, the program crashed - fixed
and several others…
sometimes in multiple files the numbering and colouring of the models were not correct - fixed;
X/UNK residues appeared as ATOM records in pdb files - fixed;
Corrections because of some problems in old PDB files (such as pdb4cpa.ent):
some obsolate atom names (AE1 etc) from old files were not recognized - fixed;
in rare cases the versions < 2.0 and ≥ 2.0 couldn't be distinguished - fixed;
now chains are always named as A,B,C..., even if the original file uses other letters, or contains unnamed chains.
shaded rings: not all rings with special bonds were filtered out - fixed;
gradient fill coloring and a few otheroptions corrected to work under Pov-Ray 3.5;
selection in the pod-file list box was not saved - fixed.
Tinker (read)
MDL's rxn and rdf (read)
Mol2Mol own format (read/write, for special occasions only)
User defined free format (read/write): by using a user-entered formatting string practically any format can be inputted or outputted (Cartesian, Z-matrix, polar) more...
Support added for PDB Fat subformat (PDBF);
Options added for residue numbering and CONECT records in PDB files;
Free format Z matrix and Cartesian input extended to Q-Chem and to some Spartan work files;
Free format extended to polar coordinates (read/write);
MXYZ reads molecule names, if present;
WinPLT: option for the automatic explicit-to-implicit conversion of hydrogens;
POV-Ray: enhanced spline algorythm; options for tightness or including all or only CA/P backbone atoms in splines.
Beilstein rosdal modified to accept ISIS generated BSD files too;
Sybyl2 format rewritten to accept files with scrambled chain and residue info;
Z-matrix generation: added manual selection of atoms;
MDL molfile to the Windows clipboard: no option dialog from now on, writes always in MDL and text formats.
Renumbering of atoms automatically by several options or manually; more...
Assigning and creating H-bonds; more...
Several new rotation and translation methods (turn selected atoms into plane, selected bond parallel with axis etc.); more...
Calculation of the bounding box of the molecule.
When removing water molecules from peptides, the residue number was not corrected – fixed;
Atom cursor was not removed at 0 position - fixed
Open file dialog: file name buffer was too small in batch input mode. Increased to accept about 500 files;
Some discrepancies in PDB file HETATM records (formatting of atom names vs. elemental symbols) have been resolved;
CACAO automatic recognition failed – fixed;
Dump all angles: two valence atoms were skipped – fixed.
Improvements in Mol2mol ver. 4.0:
(01.11.2001)
Input/output:
Graphics:
When the image is coloured by models, chains or residues, the colours used are now hold in the file "rescolors.dat" and can be edited directly or from the Preferences 3 menu;
The colouring of the molecules by models, chains or residues is now influenced by the selection mode. If nothing is selected, colour selection influences the whole image; if some part is selected, only the selected parts are coloured. The same applies when resetting the colours. Thus complicated colouring patterns can be set, which could be important when producing POV-Ray input files;
A new option appears, if a PDB file contains alternative locations. These can be coloured separately.
New features in utilities:
Backbone mode: if the chains are shown in backbone mode, the display of other molecules, ligands can be toggled on/off (mouse R-click menu);
Alternative locations in a PDB file can be deleted but a selected one;
Checking geometry: the bond distances and pyramidalities of the backbone atoms and accidental discontinuities of the chains can be automatically checked (very useful!).
adding of CH3 or CH2 centroids as dummy atoms
If a multiple file is inputted in browse mode, all of the calculations for all of the structures can be done in one step;
If the output of the results are directed to the clipboard, it can be simply pasted in tabular format into Excel.
Atom charges can be removed from the Edit menu;
Automatic addition/removal of formal charges can be disabled in Preferences 2;
Manual addition of formal charges to selected atoms.
Changes, improvements in the handling of different file types:
If the original creator program was HyperChem, aromatic bonds are erroneously assigned to amide bonds when saving in Sybyl mol2 file format. Such HyperChem produced mol2 files can be automatically corrected in Mol2Mol.
Support for the PDBQ subformat has been added (PDB files with partial atomic charges);
Support for multiple bonds and charges in the case of the RasMol/Chime subformat
Slicing/browsing of files has been extended according to models, chains or both;
If a file contains alternative atom locations, these can be separately selected and deleted.
Has been split to two subtypes, when writing. DTMM module has been upgraded according to DTMM ver. 4.
The input/output file format has been upgraded according to CACAO 98.
A few fixes were made when converting biopolymer pdb residue names to/from MacroModel format.
The newer versions of MolPic has been renamed to UltraMol. Some changes were made therefore in Mol2Mol. The earlier default file name extension *.mp3 has been changed to *.umd.
The support of Cartesian and Z matrix input has been extended to Q-Chem. All variations are allowed except Cartesian files with symbolics.
More formatting variations are supported now, including Q-Chem format;
Output of the coordinates in angstrom or nanometer units.
Support for Q-Chem added.
Options for different atom labelling has been added.
New file type: CIF/MIF file input: multiple data and bond information are supported too.
CIF: Connectivity info and bond types are now supported;
The hydrogen adding module in the previous version of Mol2Mol generated faulty atom labels, this has been fixed.
SHELX: Several small corrections; when adding automatically hydrogens, no hydrogens will be added to the Q atoms.
Far more declaration are used now, this greatly helps the manual editing and customisation of the pov files in the POV-Ray editor;
Option for writing the coordinates into a separate include file, this greatly helps the manual editing and customisation of the pov files, especially in the case of big molecules;
All of the element colours and 32 colours for the models, chains, residues are now defined in the povcolor.dat file;
The model types and cylinder widths can be separately set for the unselected and selected parts of the molecule.
Colour perspectivity is supported in the case of CPK models too;
In spline mode the splines are always coloured according to the screen; the chain must have at least three residues;
Earlier versions:
Improvements in Mol2mol ver. 3.5.0 - 3.5.1:
(02.07.2000)
Molecules-3D (m3d) read and write;
CIF write added
SHELX read and write, and special edit mode for the Q atoms;
general fractional write added;
MDL SD read and write;
MDL mol files are written in V2000 format from now on;
HyperChem hin files according to HyperChem 6.0 (support of formal charges);
Sybyl mol2
PCModel
MDL SD
Insight car
MacroModel
Xmol
MXYZ
Cambridge FDAT (multiple conversion support added too)
Cambridge MODEL (multiple conversion support added too)
Beilstein Rosdal
The new mimic pH feature is available in the case of biopolymers to protonate or not certain acidic/basic groups;
In the output file the new hydrogens will appear immediately after their parent atoms;
In the case of PDB files the correct name for the hydrogens will be derived from the name of the parent atoms;
The possible residue, chain and modul information will be preserved;
Spacefill (CPK) models can be generated as "blobs" too;
5-7 membered aromatic planar rings can be represented with circles too;
To ease the additional editing of the produced files in POV-Ray, most of the parameters are declared at the beginning of the *.pov files;
MXYZ (read and write)
Gamess type Z-matrix and Cartesian (write)
Insight car
Sybyl mol2
Macromodel
PDB
CSSR
Z-matrix input, mainly Gaussian types
Direct editing options of the povcolor.dat and *.pod files have been added to the POV-Ray window;
More colour parameters are available in the povcolor.dat file;
Tubular spline representation of peptide backbones;
Transparently shaded 3-7 membered rings;
Gradient fill colouring of the bonds is supported;
Support for the colouring according to atom charges (or other properties), residues, chains and models have been added.
This version is compiled in 32-bit mode for the Windows 95/NT and optimized for the use of Pentium processors. The 16-bit version and processors without mathematical coprocessor (486SX etc) will not be supported in the future.
ChewDraw CT (ver 4.5)
PCModel (ver 6.0)
POV-Ray (ver 3.0)
Different bond types in the POV-Ray files are available now.
The default atom colors can be changed now.
CAChe (read and write)
Gaussian Z-matrix (read and write)
MolPic (read and write)
Calculation of Verloop's QSAR steric parameters sterimol
Hide part of the molecule
Batch or multiple geometrical calculations with the aid of a script file.
MOPLO read also
Biosym_car format read and write
MDL_3.0 new format read and write
ChemDraw_CT format read and write
MacroModel format read and write
Cacao_car format read and write
MOPAC input (manual mode only)
Xmol xyz read and write
Cartesian write
POV-ray write
Showing of the peptide backbones, their conversion to a "backbone molecule" for drawing programs.
Calculation of phi, psi and omega torsional angles of peptides
Different geometrical measures of the planarity and puckering of 5-9 membered rings.
Calculation of the dipole moment (if the molfile contains atom charges)
Converting the molecule to its enantiomer.
{kind=link}
{kind=link}
{kind=link}