Main window
|
|
Main window |
|
Click the part of the Main window you want to know more about.
File menu
Open molfile:
Opens and reads a new molecule file, with the exception of X-ray and general files (M).
Open crystal:
Opens and reads an X-ray crystal file (C)
Open general:
Reads a general free format Cartesian or Z-matrix file (U). Either the file must contain the &&& or ###, GGG or $$$ markers as defined under the heading Z-matrix and Cartesian (with markers suboption), or an user defined format string had to be entered previously (user defined format suboption)
On selecting the first three items the Open file window appears. For more information about M C U, see molfiles and supported molecule files.
Preferences:
To set some general settings valid during the current and next sessions, see here
Open data file:
If instead of the atomic charges associated with the current molecule file another calculated atom property should be used, it can be done with this option. Useful mainly for browsing a calculated atom property in the graphics window or if a POV-Ray image should be colored according to this property.
Exit:
To quit Mol2Mol.
Reopen last files:
The program remembers and lists up to 15 successfully input molfiles and their types. The names of these files are added to the submenu, enabling easy and quick access, if repeated input is necessary. The suboption Remove all will clear all of the last opened files, Remove obsolete only the files that no longer exist.
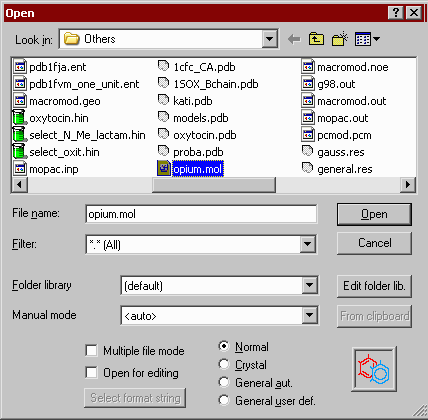
Open file
|
|
![]() To see the files only with a .mol (or any other) file name extension, type the *.mol specifier in the Molfile type box (any valid names with wildcard characters are allowed), or select from the Molfile types drop-down list.
To see the files only with a .mol (or any other) file name extension, type the *.mol specifier in the Molfile type box (any valid names with wildcard characters are allowed), or select from the Molfile types drop-down list.
![]() In the case problems with the automatic detection of the input file format you may try to use the manual mode by checking the appropriate box and selecting a file type from the appearing drop-down menu. Some newly added input file formats, which do not contain any special characteristics to reveal their type, can be reached only using this manual way (for example stand-alone MOPAC or Gaussian files).
In the case problems with the automatic detection of the input file format you may try to use the manual mode by checking the appropriate box and selecting a file type from the appearing drop-down menu. Some newly added input file formats, which do not contain any special characteristics to reveal their type, can be reached only using this manual way (for example stand-alone MOPAC or Gaussian files).
![]() Some file types may contain multiple instances of the same structure, differing only in their conformations, for example the results of a molecular dynamics experiment. In this case a multiple-to-multiple file conversion is possible by selecting this check box. Select also this option if a multiple file is intended to split into many single files (split mode)
Some file types may contain multiple instances of the same structure, differing only in their conformations, for example the results of a molecular dynamics experiment. In this case a multiple-to-multiple file conversion is possible by selecting this check box. Select also this option if a multiple file is intended to split into many single files (split mode)
![]() You may open, inspect or edit a molfile as a native text file by checking the Open for editing checkbox. Note that only small files can be edited with the very primitive Notepad (up to about 60K in Win9x). With larger files use another ASCII editor to edit the molfile. An excellent substitute of Notepad is the freeware EditPad Lite, available from http://www.editpadpro.com. You can change the default Notepad text editor from the Preferences 2 window.
You may open, inspect or edit a molfile as a native text file by checking the Open for editing checkbox. Note that only small files can be edited with the very primitive Notepad (up to about 60K in Win9x). With larger files use another ASCII editor to edit the molfile. An excellent substitute of Notepad is the freeware EditPad Lite, available from http://www.editpadpro.com. You can change the default Notepad text editor from the Preferences 2 window.
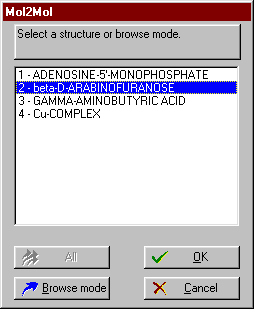
| 
|
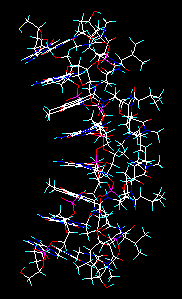 Selecting and inputting the whole DNA-actinoidin complex. |
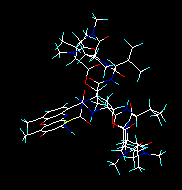 Selecting and inputting one actinoidin only. |
![]() Alternatively, the browse mode is available: the first structure in inputted, and then you can browse the others from the Graphics window.
Alternatively, the browse mode is available: the first structure in inputted, and then you can browse the others from the Graphics window. ![]() Summary of input modes
Summary of input modes
![]() If the input file contains an unknown atom type, Mol2Mol assigns a dummy atom type to it (provided its use is allowed in the molfile in question. If dummy atoms are in the input file, during the input you can choose:
If the input file contains an unknown atom type, Mol2Mol assigns a dummy atom type to it (provided its use is allowed in the molfile in question. If dummy atoms are in the input file, during the input you can choose:
a) To leave the dummy atom. In this case you have to supply the valence and the atom radius of the dummy atom (defaults: valence=2, atomradius ~ carbon);
b) Change it to another atom;
c) Remove the actual and all of the subsequent dummy atoms.
d) To read all of the dummy atoms.
![]() Input is also possible via the Windows clipboard.
Input is also possible via the Windows clipboard.
Use of the clipboard
File Open window: From clipboard button
Mol2Mol supports the direct use of the Windows clipboard. With the exception of MDL molfiles, a molfile is copied to or get from the clipboard as ASCII text. Only a few programs accept this method for moving the molecule files (for example HyperChem), therefore the usage and usefulness of this method is entirely on you and the programs you use. You may copy and paste the molfile itself to other documents etc. Maybe the most useful application of this option is the cutting of coordinates from the output of other programs (as users' general files), or cutting of molfiles from e-mails. In the latter cases use this method with caution as during the highlighting and cutting/copying from the original document blank lines can easily be added/removed to/from the beginning of the "files", and may interfere with the automatic recognition of the file type.
The From clipboard option is available only, if the data found in the clipboard is in text format or special MDL format.
If you want to use the Edit | Edit last molfile or last output file option after a clipboard operation, the Windows Clipboard viewer is called instead of the text editor.
MDL molfiles: MDL programs (ISIS) use a special connection table format when accessing the Windows clipboard. Mol2Mol will accept this format. When the To clipboard button is used, the �molfile� is placed both in the text and MDL formats into the clipboard. Nowadays several applications, such as ChemWindow, ChemDraw, ChemSketch, Chemistry 4-D Draw, HyperChem etc. supports the MDL clipboard format. The Windows Clipboard viewer cannot display data in this format!
Prior to use this clipboard format in ISIS/Draw, check the Post CTfile to clipboard option in the Preferences window of that program.
Open data file with charges or other computed data
When you input Cartesian or Z-matrix files and separate atomic charges are included in the input file, the charges can be also read in by using the @@@n marker immediately before the beginning of the atomic charges. This @@@ marker can be applied in other files as well, and not only in the case of charges. For example, a quantum chemical calculation yields to another data file, and you would like to merge these data with the molecule (inputted from another molfile) to inspect them, or to convert the molecule and these data it into a POV-ray file and colour the molecule according to this property.
As an example, find the file CACHE.CSF in the /others folder. Beside the description of the molecule it contains the results of electron density calculations: electrophilic, nucleophilic and radical susceptibilities and superdelocalizabilities. Find the beginning of these data and put the @@@n marker immediately before the beginning of the atomic charges. Substitute "n" by the column number of the desired data:
property_flags: ID N-FrontierDensity E-FrontierDensity R-FrontierDensity N-Superdelocalizability @@@3 1 0.15937 0.09829 0.12883 1.20664 2 0.15199 0.10167 0.12683 1.22933 3 0.10604 0.18979 0.14791 1.13892 ...
Only one @@@ marker may be in a file. Care must be taken that the number of data lines must be the same as the number of atoms in the current molecule. Save the file, and input it using the File | Open data file | Current molfile option (if your data happen to be in a separate file, use the Browse input option).
By moving the atom cursor in the graphics window you can inspect the electron density data (red numbers in the side bar), or you can generate a POV-Ray file with colors reflecting the relative differences in reactivity of the different parts of the molecule:
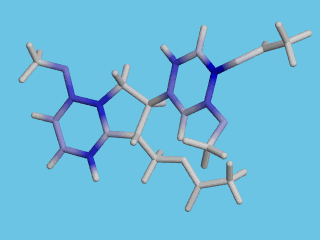
Deeper blue reflects higher electrophilic susceptibility.
Folder library
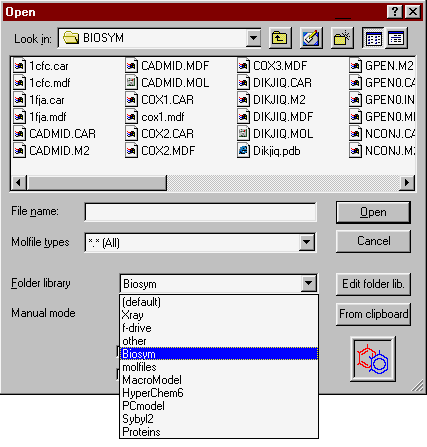
| The folder (directory) library feature provides direct and quick access (shortcut) to previously specified directories. It enables you to give each directory a descriptive name. To use it select the, then the appropriate item from the drop-down list. The Open file and Save file (Convert) windows share the same directory library. |
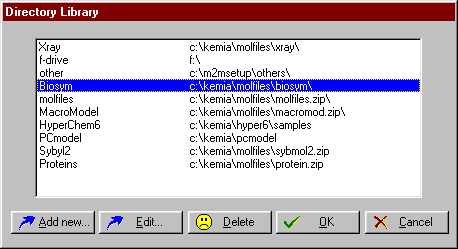
| To edit the folder library choose the Edit library button. The Edit folder library dialog window will appear. |
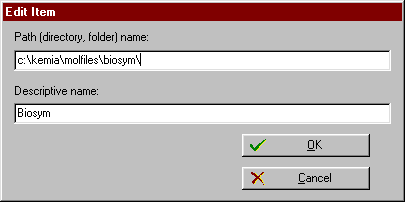
| You may add a new item, edit or delete an existing item by choosing the appropriate buttons. |
The folder library items are saved in the parent directory in the ASCII file MOL2MOL.DIR. Advanced users can edit it manually, as this information is described in the header of that file.
Edit menu
Copy graphics:
Copies the content of the opened graphics window to the Windows clipboard in bitmap format. As bitmap images are sensitive to resizing, use the appropriate options of the source or target program, if you need a better quality of picture.
Edit last molfile:
Invokes Notepad in order to edit the last molfile input (or aborted) manually. Note that only small files can be edited with Notepad (up to about 60K under Win95/98). With larger files use another ASCII editor. A very good substitute of Notepad is the freeware EditPadLite, available from http:// www.editpadpro.com. You can change the default editor in the Preferences 2 window
Show data file:
If the output of the structural (geometrical) calculations is currently directed to a file, this option becomes available and invokes Notepad in order to inspect and edit the resulting file.
Edit last output file:
Invokes Notepad in order to inspect and edit the output file of the last successful conversion.
UNIX → Windows:
The program checks automatically for unusual line endings of the input files, but this facility can be reached manually here too. In the WIndows/DOS environment each line of an ASCII text file ends with the characters cr + lf (carrige return + line feed, char(13) + char(10)). This is not the case in the UNIX or other environments. If during the file transfer from one system into another these characters are not corrected automatically (unfortunately it may happen quite often, for example if an ASCII file is moved from UNIX to Windows/DOS in binary mode), different programs will react differently: sometimes the file is automatically corrected during an input - save process, sometimes not. Mol2Mol supports UNIX files, but here the possibility is offered to correct them: the new file is written with the original name, and the original file is saved as a *.BAK file.
Clear bond types:
In rare problematic cases, when Mol2Mol cannot interpret the structure well or cannot calculate an atom type, incorrect bond types may appear: for example a double bond is placed into a bad position. Use this option to force the program to forget the actual bond types and reassign them with the next menu option or during the next upward conversion.If necessary, from the graphics window tool dialog choose the Change bond types button and add the double bonds in the critical positions. Use this option with care if special bond types (complex, weak, ionic etc) are present, because these will be changed to single ones too and may cause faulty re-assignation when using the next option.
Assign bond types:
Mol2Mol re-calculates the bond order/types, provided all of the hydrogens are present in the molecule. Complex, ionic and weak bonds cannot be regenerated add or change these manually from the graphics window tool dialog by the Change bond types button.
Generate new connections:
The present connectivities and bond orders are forgotten and new ones will be generated. This may be useful in some problematic cases.
Remove lone electron pairs:
During input, lone pairs are treated according to the Preferences settings. If lone pairs were preserved and you change your mind later, you can remove them using this option.
Remove dummy atoms:
During the input you are prompted to accept or not the dummy atoms present in a molfile. With this option all of the dummy atoms can be removed later.
Remove water molecules:
If H2O, D2O or lone O atoms are present in a (usually PDB) file, these can be removed by this option.
Remove hydrogens:
In some cases it may be useful to remove hydrogen atoms from a file, for example if you want to subsequently produce a graphical printout of a structure showing only the non-hydrogen (heavy) atoms. This option enables you to do this. (The H toggle button in the graphics window does not remove hydrogens, it only prevents them from being displayed.)
Add hydrogens:
Three suboptions are available:
The first is the normal and a very powerful option. If the molecule file does not contain the hydrogen atoms, as many of the crystallographic files and most PDB files, automatic upward conversion is not possible. You may add the hydrogens with this options. Mol2Mol is able to calculate the hybridisation state of the atoms and will add the correct amount of hydrogens in the correct geometry, using standard bond lengths. For the time being, only C, Si, Ge, Sn, O, S, Se and N atoms are taken into consideration. Mol2Mol decides according to the number of neighbours, the bond types (if available), and bond lengths. If it cannot decide, pyramidality, angles and linearity may also be taken into consideration. It must be emphasized that Mol2Mol can only calculate the atom types correctly and add the hydrogens if the structure of the input file is also correct. Inappropriate bond lengths or angles in the input file will lead to the improper assignment of atom types. If these data are not correct, the program will probably make mistakes! Unfortunately, odd bond lengths may appear even in "official" Brookhaven PDB or Cambridge CDB data files of medium or low resolution. In this situation, prior to the adding of hydrogens, in the graphics window choose the Change bond types button and add the double bonds in the critical positions, or add hydrogen atoms one by one from the graphics window. In this case the number hydrogen and the geometry is forced according to the options.
You can add hydrogen atoms ony by one to any selected atoms from the graphics window.
When adding hydrogens to lone O atoms (= structural water in peptides, DNA, etc), the positions of the hydrogens are randomized.
Limitation: For the time being, to molecules with less than 4 atoms hydrogens cannot be added.
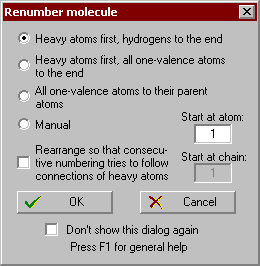
Renumber molecule:
Several options are available to renumber the molecule automatically. Alternatively, full manual renumbering is also possible.
Remove special bonds:
If the molecule contains special bond types (dummy, no-connection, unknown, coordination, ionic or weak/H-bonds), these can be removed with this option.
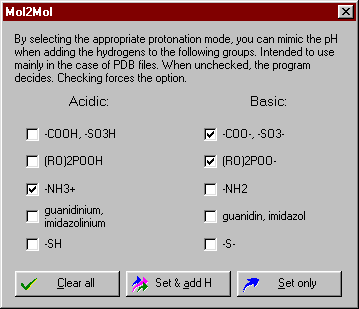
Charges:
Three suboptions are available:
3D → 2D:
Two suboptions are available:

Convert menu
This menu is only available after the successful input of a molfile.
This menu lists all the molfile formats to which files can be converted. In a few cases a subtype can be selected later from the Save file window or another dialog window. Conversion between some formats will give meaningless results, for example most conversions from two-dimensional to three-dimensional coordinates. For more details on the different file types see the molfiles section. Some modelling programs require the use of a specific filename extension for their input files (e.g. .MOL, .DAT), others only recommend a specific extension. You may add it manually, or use the default extensions option in the Save file dialog window.
If in the input phase batch or multiple mode was selected, the M letters mark that that file type can contain multiple structures/molecules.
In most cases you can do the conversion straightaway, but because of the differences in molecule files and possible errors in the geometry, especially in the interpretation of extended atom types, you may come across with several issues.
On the conversion in detail:
![]() Modelling programs can be divided into two groups. In group-1 are those that use only "natural" atom types (i.e. only atom symbols). The following modelling programs are in group-1: IISIS, Desktop Molecular Modeller, ChemDraft, CACAO, Xmol, Z-matrix, X-ray data. In addition, drawing programs such as PLUTO, PLT, ChemWindow, ChemDraw, MOPLO, WIMP and POV-Ray belong to the first group. In group-2 are programs which differentiate a large number of subtypes of the same atom, such as HC-, HO-, -COOH, -NH2, -C=C-OH, etc., or sp3, sp2, sp, >C=O, aromatic carbons, etc. If the target program belongs to group-2 during the conversion, the atom and bond types must be calculated (upward conversion), conversion in the opposite direction is more straightforward (downward conversion). The aromatic bonds are converted to alternating single and double bonds, and "query" bonds are translated into single bonds, if necessary. Mol2Mol can make most of these conversions automatically, but you can intervene if the procedure fails as a result of bad distances or in complicated (usually heteroaromatic) systems.
Modelling programs can be divided into two groups. In group-1 are those that use only "natural" atom types (i.e. only atom symbols). The following modelling programs are in group-1: IISIS, Desktop Molecular Modeller, ChemDraft, CACAO, Xmol, Z-matrix, X-ray data. In addition, drawing programs such as PLUTO, PLT, ChemWindow, ChemDraw, MOPLO, WIMP and POV-Ray belong to the first group. In group-2 are programs which differentiate a large number of subtypes of the same atom, such as HC-, HO-, -COOH, -NH2, -C=C-OH, etc., or sp3, sp2, sp, >C=O, aromatic carbons, etc. If the target program belongs to group-2 during the conversion, the atom and bond types must be calculated (upward conversion), conversion in the opposite direction is more straightforward (downward conversion). The aromatic bonds are converted to alternating single and double bonds, and "query" bonds are translated into single bonds, if necessary. Mol2Mol can make most of these conversions automatically, but you can intervene if the procedure fails as a result of bad distances or in complicated (usually heteroaromatic) systems.
![]() During an upward conversion all hydrogen atoms must be present in the structure. This restriction concerns mainly X-ray files and Protein Data Bank (PDB) files, as in many cases only the coordinates of the heavy atoms are available. If you regularly transform inorganic molecules or organic molecules which genuinely contain no hydrogens, change the default setting in Preferences | Upward conversion without hydrogens. Otherwise add hydrogen atoms with the Edit | Add hydrogens menu option.
During an upward conversion all hydrogen atoms must be present in the structure. This restriction concerns mainly X-ray files and Protein Data Bank (PDB) files, as in many cases only the coordinates of the heavy atoms are available. If you regularly transform inorganic molecules or organic molecules which genuinely contain no hydrogens, change the default setting in Preferences | Upward conversion without hydrogens. Otherwise add hydrogen atoms with the Edit | Add hydrogens menu option.
![]() If the molfile which has been input does not contain bond type information (e.g. X-ray files, PDB, DTMM, Xmol, Z-matrix files etc) and the target file type is MDL molfile, ChemDraw ct,ChemDraft, MOPLO, it is only necessary to carry out the assignment of bonds. However, the parser makes a full assignment, but the extended atom types are not taken into consideration. The different messages are not important now (but they could be useful for problematic molecules). The number of messages can be reduced by setting the Suppress conversion messages check box in Preferences.
If the molfile which has been input does not contain bond type information (e.g. X-ray files, PDB, DTMM, Xmol, Z-matrix files etc) and the target file type is MDL molfile, ChemDraw ct,ChemDraft, MOPLO, it is only necessary to carry out the assignment of bonds. However, the parser makes a full assignment, but the extended atom types are not taken into consideration. The different messages are not important now (but they could be useful for problematic molecules). The number of messages can be reduced by setting the Suppress conversion messages check box in Preferences.
![]() It must be emphasized that Mol2Mol can only calculate the atom types correctly if the structure of the input file is also correct. Inappropriate bond lengths or angles in the input file will lead to the improper assignment of atom types.
It must be emphasized that Mol2Mol can only calculate the atom types correctly if the structure of the input file is also correct. Inappropriate bond lengths or angles in the input file will lead to the improper assignment of atom types.
In the program Help several examples and tips can found.
Save file window
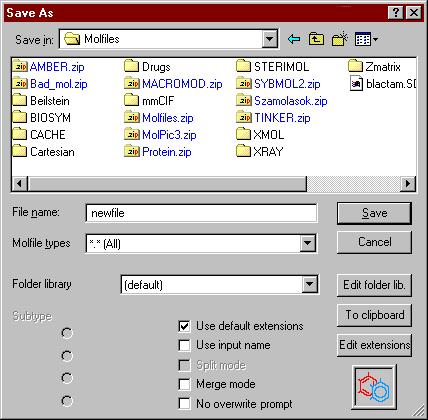
In the Save file window use the lists to find and select the drive and folder, and type (or select) the file name you would like to use, then choose Save.
![]() The Folder library feature provides direct quick access to specific directories, and you can give each directory a descriptive name.
The Folder library feature provides direct quick access to specific directories, and you can give each directory a descriptive name.
![]() The Use default extensions feature allows the automatic addition or checking of the file name extensions.
The Use default extensions feature allows the automatic addition or checking of the file name extensions.
![]() Use input name - Mol2Mol will not prompt you for a new output name, but uses the input name instead. It is advisable to switch the Use default extensions option on first, as well as to select the output directory.
Use input name - Mol2Mol will not prompt you for a new output name, but uses the input name instead. It is advisable to switch the Use default extensions option on first, as well as to select the output directory.
![]() The split (multiple-to-many) mode is available only if a multiple file was previously inputted in multiple mode: instead of writing one new file with several structures, each one will be written into a new file. Consecutive numbering will be automatically added to the supplied file name: file001.mol, file002.mol, and so on (for example a PDB file with n models to n files).
The split (multiple-to-many) mode is available only if a multiple file was previously inputted in multiple mode: instead of writing one new file with several structures, each one will be written into a new file. Consecutive numbering will be automatically added to the supplied file name: file001.mol, file002.mol, and so on (for example a PDB file with n models to n files).
![]() The merge (many-to-multiple) mode is available only, if several input files were selected (batch mode): this is an amalgamation of batch and multiple modes: after the conversion the selected input files are unified and outputted as one multiple file of several independent structures (for example n MDL molfiles to a MDL SD file with n structures).
The merge (many-to-multiple) mode is available only, if several input files were selected (batch mode): this is an amalgamation of batch and multiple modes: after the conversion the selected input files are unified and outputted as one multiple file of several independent structures (for example n MDL molfiles to a MDL SD file with n structures).
![]() Summary of input modes
Summary of input modes
You can paste the new molfile to the Windows clipboard, this can be useful in some special cases.
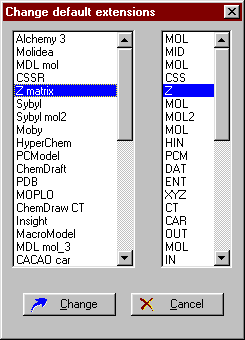
Use and edit the file name extensions library
To edit the extensions library select the Edit extensions button. A new dialog window opens, which contains the names of the different modelling programs and the corresponding extensions. Highlight the item to be edited and press Change. Enter the new extension (or "-" for nothing), then press OK. The extensions defined here appear in the file specification list box of the Open file and Save file windows.