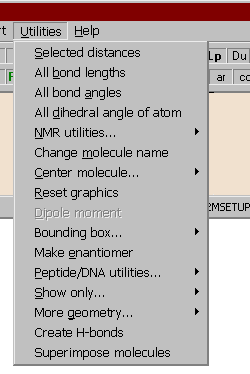
Utilities of the Main menu
This menu is only available after the successful input of a molfile.
Click the part of the menu you want to know more about.
Selected distances:
All the distances between the selected atoms are output in a triangular matrix format. Of course, the same unit of distance will be used as in the source molfile (usually the Angstrom). Click on the atoms you want to include, then press Enter.
TRIANONE.MOL
Atom-atom distances:
5-8 1.496 5-12 2.608 5-17 2.342
8-12 1.521 8-17 1.200
12-17 2.363
If you have to calculate selected distances, angles or dihedrals from a number of similar structures (in many files or in a multiple file), you may want to use the batch calculation feature.
All bond lengths:
These options enable you to output all the bond lengths of the molecule, including or excluding hydrogen atoms. To exclude hydrogens turn on the hydrogen-off button in the graphics window. If part of a molecule is selected, only the corresponding lengths will be calculated.
TRIANONE.MOL
Bond lengths:
C(2)-C(1) 1.413 C(6)-C(1) 1.406
H(20)-C(1) 1.089 C(3)-C(2) 1.400
C(7)-C(2) 1.513 C(4)-C(3) 1.406
H(21)-C(3) 1.101 C(5)-C(4) 1.413
H(22)-C(4) 1.089 C(6)-C(5) 1.400
C(8)-C(5) 1.496 H(23)-C(6) 1.092
F(9)-C(7) 1.403 F(10)-C(7) 1.368
etc
All bond angles:
These options enable you to output all the bond angles of the molecule, including or excluding hydrogen atoms. To exclude hydrogens turn on the hydrogen-off button in the graphics window. If part of a molecule is selected, only the corresponding angles will be calculated.
TRIANONE.MOLIf you have to calculate selected distances, angles or dihedrals from a number of similar structures (in many files or in a multiple file), you may want to use the batch calculation feature.Bond angles: C(2)-C(1)-C(6) 120.036° C(2)-C(1)-H(20) 119.749° C(6)-C(1)-H(20) 120.199° C(1)-C(2)-C(3) 118.932° C(1)-C(2)-C(7) 120.716° C(3)-C(2)-C(7) 120.248° C(2)-C(3)-C(4) 121.025° C(2)-C(3)-H(21) 119.066° C(4)-C(3)-H(21) 119.887° C(3)-C(4)-C(5) 120.036° C(3)-C(4)-H(22) 120.199° C(5)-C(4)-H(22) 119.750° etc
All dihedral angles of atom:
The selection of this option outputs all the starting dihedral angles belonging to a specified atom. When prompted, click on the atom around which you wish to see the dihedral angles.
TRIANONE.MOL
Dihedral angles around atom C(8):
C(8)-C(5)-C(4)-C(3): -178.34°
C(8)-C(5)-C(4)-H(22): 3.09°
C(8)-C(5)-C(6)-C(1): 178.31°
C(8)-C(5)-C(6)-H(23): -3.25°
C(8)-C(12)-C(13)-C(14): -131.32°
C(8)-C(12)-C(13)-O(18): 42.00°
In 1H NMR spectroscopy, one is often faced with the task of identifying pairs of protons which are separated by specific distances. For example, in NOE (Nuclear Overhauser Enhancement) calculations, hydrogen atoms separated by 2.0-2.5 A are particularly interesting. Moreover, in the case of methyl groups the hydrogens are averaged to a 'pseudoatom' because of the rotation, i.e. distances have to be measured from the centroid of the three hydrogen atoms. Mol2Mol provides a submenu of six options for calculating inter-hydrogen distances or adding centroids as dummy atoms:
Distances from a CH3-like pseudoatom:
This option is used for general cases. You must supply four atoms. The centroid of the first three atoms is determined, and the distance of the fourth atom from the centroid is calculated.
The program automatically locates any methyl groups in the current structure. It then searches for hydrogens within a specified radius of a chosen methyl group. The distances to hydrogen atoms are calculated from the centroid of the methyl group. The distances between the centroids of the methyl groups are also listed in the message window. The calculated pseudoatom will be represented by a red atom surrounded by a dot-surface ball. The radius of the dot-surface ball is the same as specified previously. If the graphical atom cursor is moved by the up or down buttons (or pressing U or D on the keyboard) it only jumps to the atoms which are within the specified distance from the methyl group. The calculated distances are displayed in the bottom status bar. The graphics window returns to normal by pressing the Clean and reset button in the graphical utilities toolbox of the graphics window.
Hydrogen #: Distance from Me(11): H(5) CH 2.752 H(38) C-Me(10) 2.704 Within 3.00 Å 2 hydrogen atoms were found. The distances are calculated from the centroid of the three hydrogens of the methyl group. Distance of the centroid of methyl groups from each other: C-Me(10) C-Me(11) 3.055
You are prompted to enter the maximum and minimum distances between hydrogens (in Angstroms). All hydrogen atoms within these limits are output to the message window. Methyl-methyl hydrogen distances are not included in this calculation (use the option Hydrogen distances from methyl groups for that).H-H distances between 3.00Å and 2.50Å: H(5) CH - H(7) CH 2.619 H(40) C - Me(11) 2.701 H(7) CH - H(19) NH 2.885 H(19) NH - H(24) CH 2.994 H(23) CH - H(35) CH 2.972 H(24) CH - H(35) CH 2.535 6 hydrogen pairs has been found between the given distances.
You are prompted to select a hydrogen graphically, and enter a specified radius. The specified atom will be surrounded by a dot-surface ball. If the graphical atom cursor is moved by the up or down buttons (or pressing U or D on the keyboard) it only jumps to the atoms which are within the specified distance from the hydrogen atom. The calculated distances are displayed in the bottom status bar. The graphics window returns to normal by pressing the Clean and reset button in the graphical utilities toolbox of the graphics window.
Dummy atoms are added to the molecule, positioned at the centroids of methyl groups. The atoms are bonded with dummy bonds to one of the hydrogens if unnecessary, these dummy bonds can be removed by the main menu Edit | Remove special bonds option.
Dummy atoms are added to the molecule, positioned at the centroids of methylene groups. The atoms are bonded with dummy bonds to one of the hydrogens - if unnecessary, these dummy bonds can be removed by the main menu Edit | Remove special bonds option.
Alter molecule name:
Several file formats allow the use of a name, which may also be a short comment. Use this option to change the molecule name. Note that different file formats permit names of different length.
Reset graphics:
The graphical window parameters are reset and the molecule is centered in the window in its original position (only the screen coordinates of the molecule are manipulated, not the original ones used in the conversion).
Center molecule:
Two suboptions are available:
Do not use these options if you intend to edit and save a SHELX file or when the original coordinates are important, as they will be altered!
The second option is available from the graphics window too (Center button in the side toolbar).
If the molecule contains atom charges (highlighted C in the upper status bar), the program calculates the dipole moment of the molecule.
The molecule is replaced by its mirror image.
These utilities are for displaying the backbone atoms of a peptide or DNA molecule, and for other transformations of these biomolecules:
If the peptide has a free H2N-CHR-CONH-... N-terminal, the program automatically finds it and shows the backbone. The program considers -S-S- bridges belonging to the backbone (it automatically finds them, provided they are not coupled through unusually long fragments to the main backbone).
If the peptide does not have a free N-terminal, you can enter the number of the starting N atom in the input window.
The previous two options are only for displaying the backbone, the molecule itself is not changed. This option transforms the molecule itself to a new one, stripping off all but the backbone atoms. This new backbone molecule is chemically nonsense, but you may transform it to a new file for drawing programs. After this option you cannot revert to the normal view, you have to input the molecule, if you need it again.
As before, but the new molecule contains only the alpha (CA) atoms of the peptide, or the P atoms of the DNA backbone.The simplest way to move such a molecule into a Word document: after the transformation convert it to ISIS mol format, direct the output to the clipboard. In ISIS/Draw or ChemDraw paste it into the work space, and then copy/paste into a Word document.
Resets the image to the normal view. Can't be used after Transform.
The program calculates and prints the phi, psi and omega torsional angles for each residue i of the peptide backbone:
phi: C(i)-CA(i)-N(i)-C(i-1) psi: N(i+1)-C(i)-CA(i)-N(i) omega: CA(i+1)-N(i+1)-C(i)-CA(i) N-terminal atom(s): # 1 3BRIDGES.ENT phi psi omega 1 ALA .00 -25.31 -166.66 N(1) 2 VAL -81.60 -16.26 156.97 N(13) 3 CYS -85.53 -41.35 -174.71 N(29) S1 4 ALA -53.98 -9.52 168.65 N(39) 5 VAL -99.81 -31.99 174.14 N(49) 6 SER -89.75 -.67 -173.00 N(65) 7 CYS -105.69 -17.94 171.76 N(76) S1 8 VAL -98.22 -35.15 176.95 N(86) 9 LEU -63.47 -5.21 -176.52 N(102) 10 VAL -109.45 -4.15 125.84 N(121) 11 CYS -76.10 -100.83 168.14 N(137) S2 12 GLY 70.81 -74.60 172.27 N(147) 13 GLY -82.46 -57.06 174.42 N(154) 14 CYS 18.90 -90.22 -96.97 N(161) S3 15 CYS 167.29 -89.87 144.42 N(171) S2 16 CYS 44.51 96.45 166.29 N(181) S3 17 PHE 104.63 .00 .00 N(191)where the columns are:
residue sequence number relative to the beginning of the current subunit
residue name (if any)
phi, psi and omega in degrees (±180°)
seq. number of the N atom of the current residue
S1...Sn - attachments of the nth -SS- bridge
When dumping the phi-psi-omega values of peptides, a new graph window opens containing the Ramachandran plot of phi and psi. When moving the atom cursor in the graphics window from one amino acid residue to another, a simultaneous red triangle cursor is moving in the Ramachandran plot. Use the menu bar of this auxiliary window for copying the plot to the clipboard.
If a PDB file contains more than one coordinates of the same atom(s), this option removes these but a selected one.
This is a very useful option. The bond distances and pyramidalities of the backbone atoms and accidental discontinuities of the chains can be automatically checked. Checking a peptide file and seeing the result, you may be surprised.
a) it is a true peptide file with official atom names (CA, C, N etc), orA typical output is:
b) the backbone information was previously generated by using the Backbone automatically or Backbone, mark N-terminal manually options (for example when using an xyz file).Pyramidality index of C(58) C, res LEU 6 is 0.17 (should be < 0.15) Distance of C(58) C - O(59) O, res LEU 6 is 1.150 (should be 1.21 - 1.245 Å) Distance of C(152) CA - C(153) C, res VAL 12 is 1.468 (should be 1.51 - 1.57 Å) Connectivity of C(237) C, res LEU 17 should be 3 (broken chain?)Lines in blue warn only on unusual geometry, the red ones are probably more serious mistakes in the geometry or connection.
Two suboptions are available:
The x, y and z dimensions of the bounding box of the molecule is calculated, taking into account the van der Waals radii of the atoms. The farthest extension is always the x dimension.The calculation is performed the following way:
The largest distance is looked for and then the molecule is reorientated so that the corresponding two atoms are in the x axis:The molecule is rotated around the X axis and of the resulting bounding boxes that with the minimum cross section (in the YZ plane) is accepted as the final one:
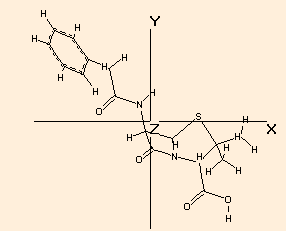
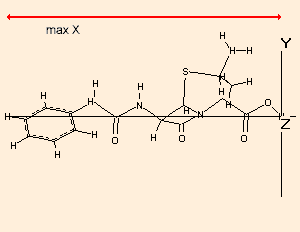
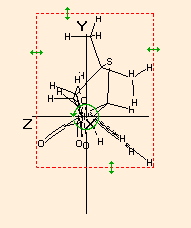
Because of the searching of the optimal arrangement, in the case of molecules with more than 13-15000 atoms the calculation may take considerable time!
This option is for the estimation of Verloop's steric parameters for QSAR (L and B1 to B5), originally calculated by the program Sterimol. These parameters characterize the shape of a molecule or substituent, the geometrical features which might significantly influence the biological activity. L is the length of the molecule/substituent, B1-B4 is the measure of the width in four rectangular directions, B5 is the maximum width perpendicular to L. For a detailed description of these parameters consult :
Verloop, A., Hoogenstraaten, W., Tipker, J.: Development and Application of New Steric Substituent Parameters in Drug Design in: Drug Design (E.J. Äriens, ed), vol VII., chapter 4, Academic Press, N.Y.,1976
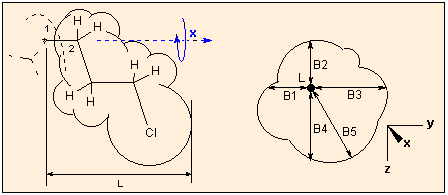
When calculating these parameters, the molecule is regarded as a substituent: you are prompted to select a first atom (usually a hydrogen) from the graphics window, this plays the role of the atom of the parent phantom molecule to which the substituent is connected, and a second atom: this is the joining atom of the substituent. The molecule is reoriented so that the first atom is in the origo (0,0,0), and the second atom lies in the X axis. The graphics window will show this. The farthest extension in the X direction of the molecule is calculated as L, the atoms are represented as spheres with their van der Waals radii. Next the "substituent" is rotated around the X axis (atoms #1-#2). During the rotation the minimum and maximum distances to the X axis in the XY and YZ planes are calculated. When the absolute minimum is found, it will be B1, and the corresponding other three distance values, in ascending order, B2 - B3; the absolute maximum is taken as B5.
With this menu option part of a complex molecule can be hidden. When selecting an atom with the mouse, the graphics window displays only the atoms within a "distance" of 4 or 6 bonds from the selected atom, or within a given radius. All other atoms are hidden. Use the reset option to reverse this command and return to normal view.
If you have to calculate different geometrical data of the same molecule many times, for example you have a molecule in many different conformations written as several one-molecule molfiles or as a multiple molfile (as the example file MACROMOD.OUT), this option can help very much. You define the data to be calculated in a small script file and Mol2Mol calculates the data without searching and clicking the atoms in the graphics window.
Two types of calculations are available:
This is for calculating the distances of given atom pairs, comparing it with given distances (for example with measured NMR NOE data), and an RMS value will be calculated from the differences of the given and calculated data. For example, you have many conformations resulting from a molecular dynamics or Monte-Carlo simulation,. and you want to compare distances with those of obtained from NOE experiments.
Prepare a data file with Notepad or any other ASCII text editor as follows:
a) The name of the data file must be the same as the molfile, but with the extension *.NOE (e.g. myfile.out - myfile.noe ) and must reside in the same subdirectory (folder).
b) The first lines beginning with the character ; may contain any remarks.
c) Each of the subsequent lines contain the number of the two atoms followed by the given experimental distance (see MACROMOD.NOE in the \others subdirectory of Mol2Mol):
;sample file for macromod.out 87 70 1.92 95 88 2.22 36 55 2.40 103 78 2.26 102 78 3.23 87 72 2.91 87 71 3.42 ...After the input of the structure file (for example the sample file MACROMOD.OUT in the \others subdirectory) click on the Utilities | More geometry | Compare distances option, the result will be:
CONF 43 E = -4.09 sample file for macromod.out atom 1 2 meas. calc. delta delta% 87 70 1.920 2.193 0.273 14.237 95 88 2.220 2.200 -0.020 -0.912 36 55 2.400 2.307 -0.093 -3.876 103 78 2.260 2.204 -0.056 -2.492 102 78 3.230 3.504 0.274 8.474 87 72 2.910 4.073 1.163 39.983 87 71 3.420 4.517 1.097 32.086 . . . 56 33 3.410 3.567 0.157 4.607 103 29 4.230 5.300 1.070 25.298 103 30 4.230 5.678 1.448 34.233 RMS = 4.305where each line contains the number of the two atoms, the input distance and the calculated distance between the two atoms, the difference and the difference %. The RMS value at the end is calculated as SQRT(SUM (dcalc. - dmeas.)2).
This option is for the multiple calculation of distances, angles and dihedral angles. Prepare a data file with Notepad or any other ASCII text editor as follows:
a) The name of the data file must be the same as the molfile, but with the extension *.GEO (e.g. myfile.out - myfile.geo ) and must reside in the same subdirectory (folder).
b) The first lines beginning with the character ; may contain any remarks.
c) The data lines may contain two, three or four atom numbers. The program will interpret these as input data for distance, angle or dihedral angle calculation. After the last numerical data an optional short text may appear, these will be appended to the calculated data (see MACROMOD.GEO in the \others subdirectory of Mol2Mol):;data file for Macromod.out. 87 70 32 31 34 32 31 34 45 psi Ala1 44 46 49 50 phi iGlu 46 49 50 51 psi iGlu 50 51 43 61 phi Dap 51 43 61 82 psi Dap 61 82 81 84 phi Ala4 82 81 84 79 psi Ala4 84 79 68 92 phi Ala5After the input of the structure file (for example the sample file MACROMOD.OUT in the \others subdirectory) click on the Utilities | More geometry | More geometry option, the result will be:
CONF 43 E = -4.09 data file for Macromod.out DIST 2.193 H(87)-H(70) PIM ANG 111.3 N(32)-C(31)-C(34) ALA DIH -13.6 N(32)-C(31)-C(34)-N(45) psi Ala1 ALA DIH 57.5 C(44)-C(46)-C(49)-C(50) phi iGlu D_IG DIH -126.5 C(46)-C(49)-C(50)-N(51) psi iGlu D_IG DIH -126.8 C(50)-N(51)-C(43)-C(61) phi Dap PIM DIH 89.5 N(51)-C(43)-C(61)-N(82) psi Dap PIM DIH 156.4 C(61)-N(82)-C(81)-C(84) phi Ala4 DA DIH -131.6 N(82)-C(81)-C(84)-N(79) psi Ala4 DA DIH 158.5 C(84)-N(79)-C(68)-C(92) phi Ala5 DAIn the case of biopolymers, if the input file contains residue/subunit names (amino acids etc), these will be appended to the end of the lines.
If you redirect the results of these calculations into a file, you can export them easily into MS Excel or any other Windows application.
In HyperChem you may create named selections. These are saved with the molecules. On selecting this menu option the corresponding geometrical values of all two, three or four atom selections will be dumped:DIST dist1 1.478Å N(3)-C(4) DIH tor3 170.742° C(1)-C(2)-N(3)-C(6) ANG AB-angle 114.893° H(14)-N(7)-C(1) DIST LINE 1.478Å C(4)-N(3)This feature works in the case HCS (HyperChem conformation search) files and in browse mode as well.
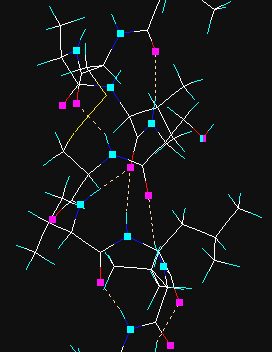
Hydrogen bonds are searched for and created. In the first step hydrogen donor and acceptor atoms are assigned and then if certain geometrical criteria are satisfied, the new H-bond is added. In vector view mode the donor and acceptor atoms are displayed as cyan and magenta rectangles, the H-bonds as yellow dotted lines. Prior to the use of this method all hydrogen atoms must be present, otherwise add hydrogens via the Main | Add hydrogens menu option.
The cyan and magenta markings can be removed with Clear button in the graphics tool dialog. If the hydrogen atoms are removed later, hydrogen bonds will jump and sprout from the donor atoms.
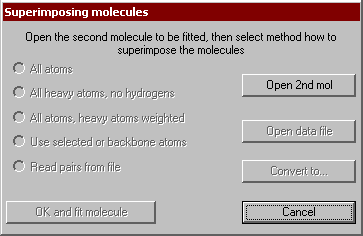
This utility superimposes all or specified atoms of two molecules. The current molecule is regarded to the template one and its orientation is kept fixed, while the 2nd fitted molecule is translated and rotated to minimize the difference of the distances of the corresponding atom pairs. The pairs of atoms to be fitted may have assigned weights, a larger weight will force a tighter fit for the atom pair.
At first you are prompted to select the second molecule. If the number of atoms are not same in the two molecules, a warning appears. Next you have to select how the program selects the corresponding atom pairs and what weighting method is used.
- All atoms are selected with equal weighting (1 for each atom);
- All heavy atoms are selected, no hydrogens are regarded;
- All atoms are considered, the weight factor is 2 for heavy atoms, 1 for hydrogens;
- The atoms previously selected from the graphics window are considered only (residues, chains, or a peptide/DNS backbone).
It is entirely your responsibility that the atoms structurally correspond to each other: 1- 1, 2 2, n m. Otherwise the result will be nonsense. If n ≠ m only the smaller number of atoms are considered.- The program prompts for the name of a script file which contains the data of the atom pairs and the weights. It is a simple free format ascii text file and must have the following structure:
4 8 12 1 9 13 1 15 16 2 17 22 2
The text window outputs the results of the fitting procedure in the following format (it can be directed to a file or to the clipboard, too):Template mol: E:\M2MSETUP\OTHERS\MOLECULE1.PDB Fitted mol: E:\M2mSetup\Others\MOLECULE2.PDB Mol templ Mol fit dist weight * C(1) C(1) 0.384 1 * C(2) C(2) 0.390 1 * O(3) O(3) 0.407 1 * C(4) C(4) 0.488 1 * S(5) S(5) 1.906 1 H(6) H(6) 2.008 0 H(7) H(7) 1.566 0 H(8) H(8) 1.151 0 H(9) H(9) 0.589 0 * N(12) N(12) 0.431 1 * C(13) C(13) 0.362 1 ... Weighted rms deviation of the selected 68 atom pairs: 0.582 Simple rms deviation of all 133 atom pairs: 0.971The asterisks mark the atom pairs used in the calculation ( = atoms with not zero weight). If the molecules are peptides and the file contains secondary structural info (helix, sheet, turn), the rms calculations are performed for these corresponding atoms too:Weighted rms deviation of the selected 951 atom pairs: 1.474 Weighted rms deviation of the 228 HELIX atom pairs: 0.995 Weighted rms deviation of the 384 SHEET atom pairs: 0.962 Weighted rms deviation of the 133 TURN atom pairs: 1.312 Weighted rms deviation of the 206 other atom pairs: 2.470 Simple rms deviation of all 1196 atom pairs: 1.888Summary: Structure rms sel. rms all 1 1.848 1.848 2 1.668 1.668 3 1.696 1.696 4 1.475 1.475 ... 29 1.811 1.811 30 1.623 1.623 31 1.783 1.783
+ 
+... =