Examples
If no hydrogens are present in the molecule, do this way.
Example 1
 1) Read the protein file KATI.PDB and open the graphics window. This is a small cyclopeptide containing only part of the hydrogen atoms. The file originates from an NMR experiment (conformation elucidation with NOE). The creator program is unknown (probably an NMR utility program).
1) Read the protein file KATI.PDB and open the graphics window. This is a small cyclopeptide containing only part of the hydrogen atoms. The file originates from an NMR experiment (conformation elucidation with NOE). The creator program is unknown (probably an NMR utility program).
2) There will be no problem with the conversion if a simple molfile like Z-matrix, Cartesian, CSSR, Xmol or a similar one is to be produced. If the transformation is upwards, the exact atom and bond types will also be needed: when you try to convert the molecule to Alchemy, Sybyl, Insight or HyperChem etc. formats, several warnings appear in the message window, because where the hydrogens are absent, the parser cannot calculate the atom and bond types!
3) As a solution, select the Edit | Add hydrogens | as atoms option. When all hydrogens are present in the molecule, the conversion will proceed without problems.
Example 2
 1) Read the last molecule from the sample file GENERAL.RES, it resides in the subdirectory OTHERS. Use the File | Open general | automatically option, as this is a general free format Cartesian or Z-matrix file without bond information. Answer twice “no” to the pop-up questions. This file contains a molecule with an oxime methyl ether group in which the distances are incorrect.
1) Read the last molecule from the sample file GENERAL.RES, it resides in the subdirectory OTHERS. Use the File | Open general | automatically option, as this is a general free format Cartesian or Z-matrix file without bond information. Answer twice “no” to the pop-up questions. This file contains a molecule with an oxime methyl ether group in which the distances are incorrect.
2) In the graphics window you will see that two bonds are missing. Activate the Alter connections or bond types tool bar via the graphical utilities dialog window, and add two single bonds: the missing C7-O6 and the C3-N5 bond of the oxime moiety: first of all select the button 1 from the side toolbar, and then click on the appropriate atoms.
3) Now convert the molecule to an Alchemy file. The program displays two messages in the text window, as the parser cannot interpret the structure correctly:
N(5) could not be exactly recognized4) Input the molecule again, but this time when adding the missing bonds, create a double bond between the C3 and N5 atoms (Answer “Some” for the question of the appearing message box). The conversion will now proceed without problems.
C(3) >C(+)– carbonium
Example 3
A
1) In the Open file dialog select the file PDB1FJA.ENT from the OTHERS folder. As Mol2Mol realizes that this is a PDB file containing several models, it opens a list box and prompts you to select & input one or more models.
2) Click on the first model only and press the OK button. Mol2Mol will locate it and open the same list box, as the model contains four different structures (two DNA chains with two intercalated actinoidine molecules).
3)
| If you select all of the four structures, the whole assembly (all of the four molecules) will be inputted: | If you select the third structure, one of the actinoidine molecules will be inputted: |
|
|

|
| Subsequent conversion results in a file containing all of the structures. In a new PDB, Sybyl mol2, Insight, Macromodel or HyperChem file the structural info about the four subunits will be preserved. | The output file of any subsequent conversion will contain this structure only. |
Alternatively you may select Browse mode in the second dialog. You can inspect the four molecules one by one by using the green arrow buttons in the graphics window. Note the differences without/with the Fix image size button activated.
B
 1) Input the file again as above.
1) Input the file again as above.
2) When you are prompted, press now the All button. The whole file will be inputted. As the indicators in the upper status bar show, the work place now contains 3 models (“molecules”), 12 chains and 102 residues altogether.
3) Subsequent conversion to Sybyl mol2, Insight or Macromodel files will lead to a similar file. In the case of HyperChem the "model" information will be lost, but chains and residues will be preserved. In the case of PCModel, Xmol, MXYZ and MDL sdf files three separate “molecules” will be put into the new file but without chain and residue information (in multiple mode, otherwise the output file will contain everything as one molecule).
All other output file types will contain everything as one molecule.
4) If you repeat the steps 1-3, but in the Open file window you set the Multiple mode check box and in the Save file window the Split mode check box, the three models will be outputted into file001, file002 and file003.
Example 4
This and the next example show show to check the geometry of a peptide and to manipulate a crowded image to inspect the exact site.
1) Input the sample file 1SOX_Bchain.PDB
2) Run from the main menu the Utilities | Peptide/DNA chain...| Check peptide geometry option. The backbone of the peptide is checked and the following report is displayed in the text window:
Connectivity of C(659) C, res ASP 83 should be 3 (broken chain?) Distance of C(659) C - O(660) O, res ASP 83 is 1.253 Ĺ (should be 1.21 - 1.245) Distance of C(822) CA - C(823) C, res LYS 105 is 1.493 Ĺ (should be 1.51 - 1.57) Distance of C(823) C - N(830) N, res LYS 105 is 1.304 Ĺ (should be 1.32 - 1.35) Distance of C(1292) C - O(1293) O, res SER 164 is 1.252 Ĺ (should be 1.21 - 1.245) Pyramidality index of C(1298) C, res ARG 165 is 0.16 (should be < 0.15) Distance of C(1480) C - N(1489) N, res ARG 187 is 1.306 Ĺ (should be 1.32 - 1.35) Distance of C(1998) CA - C(1999) C, res SER 255 is 1.500 Ĺ (should be 1.51 - 1.57) Pyramidality index of C(2087) C, res ASN 267 is 0.41 (should be < 0.15) Distance of C(2087) C - O(2088) O, res ASN 267 is 1.410 Ĺ (should be 1.21 - 1.245) Distance of C(2148) C - O(2149) O, res ASP 274 is 1.251 Ĺ (should be 1.21 - 1.245) Distance of C(2165) CA - N(2164) N, res GLY 276 is 1.419 Ĺ (should be 1.44 - 1.49) Distance of C(2166) C - N(2168) N, res GLY 276 is 1.307 Ĺ (should be 1.32 - 1.35) Distance of C(2170) C - N(2179) N, res PHE 277 is 1.289 Ĺ (should be 1.32 - 1.35) Distance of C(2261) C - N(2270) N, res ARG 290 is 1.304 Ĺ (should be 1.32 - 1.35) Distance of C(2506) C - N(2511) N, res PRO 319 is 1.306 Ĺ (should be 1.32 - 1.35) Distance of C(2659) C - N(2664) N, res PRO 338 is 1.361 Ĺ (should be 1.32 - 1.35) Distance of C(2706) CA - C(2707) C, res GLN 345 is 1.494 Ĺ (should be 1.51 - 1.57) Distance of C(2707) C - N(2714) N, res GLN 345 is 1.300 Ĺ (should be 1.32 - 1.35) Distance of C(2961) CA - C(2962) C, res GLY 380 is 1.500 Ĺ (should be 1.51 - 1.57) . .
| The blue lines draw your attention only to some minor deviances from the average geometry, but the red lines indicate more serious errors. The default image is very crowded, therefore: 3) In the left toolbar click the Jump button, enter the atom number of the first mistake (659) in the input box, press OK. 4) Press the Center button in the toolbar |

|
| 5) Zoom-in the image by pressing simultaneously the shift button and the mouse L-button, and dragging upward. The result should look as shown here. You can see that the peptide chain ends suddenly at ASP 83. | 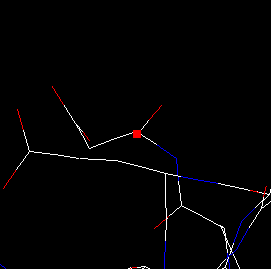
|
| Decrease somewhat the size of the image and move the cursor upwards with the red arrow button, until it reaches the N of the next residue, ASP 84. If you measure the distance of N(665) and C(659), it is 17.3 Å! | 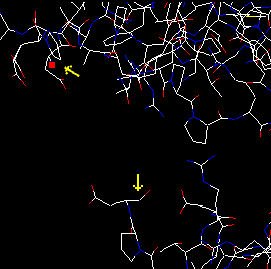
|
| 7) Now have a look at the second mistake at C(2087). Repeat the steps 2-3-4, then rotate the image so the atom with the cursor on is not obscured by any other atoms. 8) From the main menu select the Utilities | Show only…| within 6 atoms option, and when prompting, click again on atom C(2087). Only the immediate surrounding of the atom is displayed. On rotating the image you may see that the C-O bond is too long and the peptide bond is not planar. 9) You may select and check any other atoms by repeating the steps 3 and 4. You will notice that when the Show only feature is activate, pressing the Center button not only centers the image at the current atom, but visible part of the molecule follows the change as well. Similarly, the Jump button brings to the center the appropriate atom and displays only its sorrounding. This is a very handy feature to inspect big molecules and for example to step along the backbone of a macromolecules, as shown in the next example. |
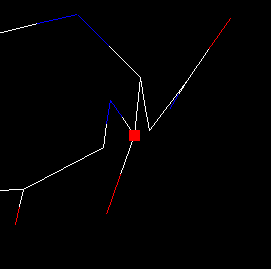
|
Example 5
This example shows how to step along the backbone of a peptide:
|
|
|
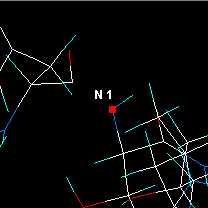
| Jump to atom #1 and center.
|
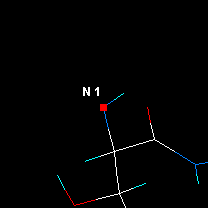
Zoom in.
|
Select Utilities | Show only within 6 atoms.
|
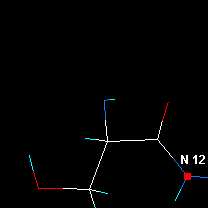
|
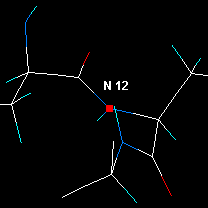
|
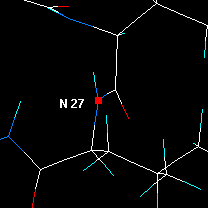
|
| Check distances etc, then click on the next N atom. |
Press Center. Now the visible area is around the next residue. |
Click on the next N and press Center... and so forth. |
Example 6
This example shows the procedure of inputting coordinates from files with unsupported formats, from an e-mail etc. using the User defined free format method.
Let's suppose we have a file with the following coordinate section:
… … 2-methyl-aurenon 23 45 1 0x0002 –0.1326 0.3333 1.2345 charge 0.1234 CA C 2 0x0002 0.8765 0.5432 1.3232 charge 0.0023 CB C 3 0x0002 1.1212 0.3560 2.0002 charge –0.3434 O1 O …The program has to interpret only the data columns in colour:
1 0x0002 –0.1326 0.3333 1.2345 charge 0.1234 CA Cnamely the three coordinates, the partial charge and the elemental symbol.
 1) Select exactly the lines with the coordinates and copy them to the Windows clipboard.
1) Select exactly the lines with the coordinates and copy them to the Windows clipboard.
2) Open the File | Preferences 3 option dialog, press the Input format strings button and add the new input format string: d d x y z d p d e
(it means: skip 2 data colums, read x data, read y data, read z data, skip next data, read partial charges, skip next data, read elemental symbols.)
Ensure before pressing the OK button that the newly entered format string is selected. The program remebers the last selected format string, therefore this step should be omitted if coordinates with the same format are to be inputted.
3) From the main menu select File | Open general | user defined format.
4) In the File open dialog click on the From clipboard button.
