Graphics window
|
|
Graphics window |
|
Click the part of the Graphics window you want to know more about.


Atom-atom distances:
You are prompted to select two atoms. The distance of these atoms will be displayed in the message window and the side tool bar.
Distances to nearest neighbours:
In the bottom status line the distances to the nearest neighbour atoms appear automatically. The default value of the distance limit is 1.8 Angstrom. You can change it in the second Preferences window. Yellow bonds will also emphasize these atoms (this does not apply when the axes are drawn)
Bond angles:
The bond angle defined by three atoms is calculated. The angle between these atoms will be displayed in the message window and the side tool bar.
Dihedral angle:
The dihedral angle as defined by four atoms is calculated. The dihedral angle will be displayed in the message window and the side tool bar.
Dihedral angle of two planes/ring:

The program prompts you to define the two planes by specifying three or more atoms in each. The best fitted mean planes and their angle are calculated. The two planes may contain common atoms (e.g. two spiro rings). As two planes define two angles, one obtuse and one acute (the sum being 180 degrees), the program calculates both and displays them in the message window. (Previous Mol2Mol versions used exact planes defined by three atoms.)
Distance from a least-square plane/centroid:
Select at least three atoms to define the plane. Press Enter after marking the last atom. Finally, click on the base atom outside the plane. Mol2Mol provides two approaches to this calculation. Each has advantages for different types of structure. Select whichever is more appropriate for the current structure:
a) A plane is fitted to the selected group of atoms by a least-squares procedure, and the distance of the base atom from this plane is calculated.
b) The centroid of the atom group is calculated, and the distance of the base atom from this centroid is determined.The best fit plane is shown in yellow. It remains visible until the button is released. 

Pyramidality:
You are prompted to enter the numbers of the three atoms of the base plane, followed by the out-of-plane atom. Pyramidality has several definitions, therefore four different indicators are calculated:

a) The distance d of the out-of-plane atom from the plane defined by the three other atoms (altitude of the pyramid). Note that the altitude of the pyramid depends on the bond lengths, so this is not an exact measure of pyramidality.

b) The deviation of the sum of the three angles around the central atom from 360 degrees is independent of bond lengths. This measure is insensitive to small pyramidality.
c) A new definition of pyramidality is index P, as described by Radhakrishnan and Agranat (Struct. Chem. 2, 107-115 (1991)). The magnitude of P varies between 0 and 1.0 (e.g. it is 0.77 for nitrogen in ammonia with bond angles of 109.4 degrees), and is positive, if viewed from the central out-of-plane atom and the order of the other atoms is clockwise. P is valid only if the bond angles are 90° < Q < 180°.
d) The pi-orbital axis vector (POAV) defined by Haddon (J. Amer. Chem. Soc. 112, 3385 (1990) is set to have equal angles with the three sigma bonds. This angle theta (or 90-theta) is also a good measure of deviation from planarity. It is 90° for planar systems, 101.04° for the ball-shaped buckminsterfullerene and 108.9° for a tetrahedral carbon atom.
Planarity and puckering of rings:

Click on an atom of a 5-9 membered ring (if the atom belongs to more than one ring, the program prompts to click on another atom to identify the ring). The program calculates a best fit plain to the atoms, as well as the following geometrical data:
Geometric and puckering data of the selected ring: Atom: Distance from the Distance if the actual atom is least-square plane: excluded from the plane: C(9) -0.280 Å. max -0.676 Å. S(8) 0.276 Å. max 0.844 Å. max C(4) -0.239 Å. -0.588 Å. N(1) 0.091 Å. min 0.207 Å. min C(12) 0.151 Å. 0.382 Å. SUM dev^2: 0.493 Ring dihedral angles: C(9) S(8) C(4) N(1) 36.600° S(8) C(4) N(1) C(12) -26.803° C(4) N(1) C(12) C(9) -3.105° N(1) C(12) C(9) S(8) 31.218° C(12) C(9) S(8) C(4) -41.015° The sum of the ring dihedral angles is -3.104°The distance of each atoms of the ring from the best fit plane is calculated by two methods: the current atom itself is included in the calculation of the best fit plane, or not. The atoms having the maximum and minimum amplitude (distance from the plane) are marked by max and min. The sum of the squared distances from the ideal plane and their average are also calculated, as measures of ring puckering. Finally, the endocyclic torsion angles of the ring atoms are also calculated.
The best fit plane is shown in yellow. It remains visible until the button is released (manually or automatically, as set in Preferences).
Remove atom:
Use this option to remove an atom from the molecule. The program will prompt for the atom to be selected. You can use this option for instance to remove dummy atoms from a molecule, if they were not automatically removed during the input.
Change atom:
On clicking an atom, you will be prompted to enter the new atom symbol. If the current molfile uses extended atom types (Alchemy, Sybyl, Hyperchem, PCmodel, Moby, MacroModel, Cache), you will have to supply the extended atom types as well. These atom types will be checked to see whether they are valid in the current molfile. If they are not valid, a warning will appear asking whether the change is to be accepted or rejected.
Change bond connections or types:
If you select the Change bond type button from the upper menu bar, the side bar will be replaced by another set of buttons to enable the selection of the appropriate bond type. First select the bond type (1-2-3, 0 to clear the bond, ar for aromatic, am for amide, du for dummy, unk for unknown, no for no-connection, co for coordinative, io for ionic and w for weak), then click on the two ends of the bond. Only use bond types, which are allowed in the target file type.
When a crystal coordinate, Cartesian or Z matrix file is loaded into the program, Mol2Mol generates a connectivity table for it. If the file contains inaccurate geometry, the program may generate "dummy" bonds between the atoms, or some necessary bonds will not appear. If this happens, use this option to add or remove bonds, or change the bond types.
The screen will be re-drawn after changing the bond type, when the button is released. Alternatively, press any button which forces a screen re-draw.
Add hydrogen atoms:
While with the main Edit | Add hydrogens option you can add all of the hydrogen atoms to a molecule at once, this option adds the hydrogen(s) only to the selected atom. In automatic mode, Mol2Mol determines automatically the number of hydrogens and the geometry of the newly created center. When you select another button from the side menu bar, the program enters into force mode: the atom and bond types, bond lengths are not taken into consideration, but some necessary basic geometric conditions.
Add formal charges:
Use this option to add/remove formal charges to/from atoms. You are prompted to click on an atom, then right-click and select the appropriate formal charge (-3 +3).
Colouring and selecting parts of the molecule or the assembly:
|
|

|
On residues, subunits, models and multiple files:

![]() The majority of molecule files describe really one molecule only (even if this is for example a coordination complex). Some molfiles may contain more tahn one molecules, like PCModel, Xmol, MXYZ and certain crystal structure files. These molecules may be completely different, or they may contain for example different conformers of the same structure.
The majority of molecule files describe really one molecule only (even if this is for example a coordination complex). Some molfiles may contain more tahn one molecules, like PCModel, Xmol, MXYZ and certain crystal structure files. These molecules may be completely different, or they may contain for example different conformers of the same structure.
![]() More advanced files possess multiple levels of structural information: residues in biopolymers (like amino acids in the proteins, nucleosides in the nucleic acids or other embedded moieties) represent the first (lowest) level. Residues are simple to treat, because all of the programs supporting residue information handle these elements in the same manner. Of the supported molfile formats PDB, Sybyl mol2, Insight car, HyperChem hin, MacroModel and SHELX files contain residue information. When interconverting between these formats, this information will be preserved and written into the new file.
More advanced files possess multiple levels of structural information: residues in biopolymers (like amino acids in the proteins, nucleosides in the nucleic acids or other embedded moieties) represent the first (lowest) level. Residues are simple to treat, because all of the programs supporting residue information handle these elements in the same manner. Of the supported molfile formats PDB, Sybyl mol2, Insight car, HyperChem hin, MacroModel and SHELX files contain residue information. When interconverting between these formats, this information will be preserved and written into the new file.
![]() In fact, a peptide or nucleic acid is often an assembly of several molecules called chains. PDB, Sybyl mol2, Insight and MacroModel support chain information. In HyperChem chains are treated as independent molecules.
In fact, a peptide or nucleic acid is often an assembly of several molecules called chains. PDB, Sybyl mol2, Insight and MacroModel support chain information. In HyperChem chains are treated as independent molecules.
![]() Finally, these advanced molfiles may contain several molecules (meaning now the whole assembly, such as a peptide or nucleic acid plus water molecules, ligands etc). In the case of PDB files these are named models. Such files are referred to as multiple files throughout this document. When using PDB files it is assumed that models have the same primary structure, differing only in their conformation (as a result of MD experiments or structure determination by NMR). In Sybyl mol2, Insight and MacroModel they can be totally different structures too. To the contrary PCModel, Xmol, MXYZ, SDF and several X-ray files contain only this utmost level info, but no residue or chain information.
Finally, these advanced molfiles may contain several molecules (meaning now the whole assembly, such as a peptide or nucleic acid plus water molecules, ligands etc). In the case of PDB files these are named models. Such files are referred to as multiple files throughout this document. When using PDB files it is assumed that models have the same primary structure, differing only in their conformation (as a result of MD experiments or structure determination by NMR). In Sybyl mol2, Insight and MacroModel they can be totally different structures too. To the contrary PCModel, Xmol, MXYZ, SDF and several X-ray files contain only this utmost level info, but no residue or chain information.
![]() As a consequence, this multilevel structural information can only be moved into a new file format between Sybyl, Insight, MacroModel and PDB. When transforming into HyperChem format, the utmost level cannot be preserved, and when converting into PCModel, Xmol and MXYZ xyz files the chain and residue information will be lost.
As a consequence, this multilevel structural information can only be moved into a new file format between Sybyl, Insight, MacroModel and PDB. When transforming into HyperChem format, the utmost level cannot be preserved, and when converting into PCModel, Xmol and MXYZ xyz files the chain and residue information will be lost.
![]() To make things more intricate, the same structure (assembly) will not be treated on the same way by different programs, for example the structural water in proteins: when writing a structure in PDB file format, water molecules are formally treated by some programs as one chain consisting of several water "residues", by other programs as independent chains or molecules.
To make things more intricate, the same structure (assembly) will not be treated on the same way by different programs, for example the structural water in proteins: when writing a structure in PDB file format, water molecules are formally treated by some programs as one chain consisting of several water "residues", by other programs as independent chains or molecules.
Mol2mol offers several ways for inputting multiple structural files and convert the whole file or only part of it into multilevel format or "all into one". Experiment with the sample files in the mol2mol/other folder:
pdb1fja.ent PDB file of actinomycin DNA complex, with three models 1cfc_ca.pdb PDB file with ten models, contains only the CA backbone atoms oxytocin.pdb oxytocin molecule in PDB format oxytocin.hin oxytocin molecule in HyperChem format macromodel.out MacroModel file with three conformers of an adamantoyl glycopeptide pcmodel.sst PCModel file with four different componds
Multiple file conversion
Some file types (Brookhaven PDB, Sybyl mol2, MacroModel, Insight car, PCModel, Xmol, MXYZ) may contain multiple instances of the same structure, differing only in their conformations, for example the results of a molecular dynamics experiment. In this case a multiple-to-multiple or multiple-to-many file conversion is possible by selecting this check box of the Open file window. After inputting the first structure and converting it to the desired file, the forthcoming structures will be read, converted and appended automatically.

|
")
|
Molecule file with multiple instances of the same structure. Multiple conversion results in a similar file. As the indicators in the upper status bar show, the workplace now contains 3 models (molecules), 12 chains and 102 residues altogether.
|
Input and conversion of one structure only. |
Among the supported file types Brookhaven PDB files signal only clearly this type multiplicity: the presence of MODEL and ENDMDL tokens means that the same structure is present in multiple instances. In this case you will be prompted to read in only the first structure or the whole file. If you select the latter similar multi-structure output files will be automatically produced during the conversion in the case of Sybyl, MacroModel and Insight types. Conversion to Xmol xyz, Mxyz and PCModel results in a similar multiple file, but without the residue and chain information. In the case of HyperChem model information is lost, but residue and chain info is preserved.
Summary of different input and output modes
![]() The basic and most used mode is to select simply a molecule file from the input window. The file is inputted and is ready for further manipulations.
The basic and most used mode is to select simply a molecule file from the input window. The file is inputted and is ready for further manipulations.
|
|

|
|
|

|

![]() If the All option is available, you may input all or select only several of the molecules.
If the All option is available, you may input all or select only several of the molecules.
![]() Otherwise you can select and input one molecule at a time.
Otherwise you can select and input one molecule at a time.
![]() Alternatively, the browse mode is available: the first structure in inputted, and then you can browse the others from the Graphics window (the Data button is available only in the case MDL sdf or rdf files and toggles the display of associated data in the text window).
Alternatively, the browse mode is available: the first structure in inputted, and then you can browse the others from the Graphics window (the Data button is available only in the case MDL sdf or rdf files and toggles the display of associated data in the text window).
![]()
![]() Multiple-to-multiple mode: an input file with several structures will be transformed into another multiple structure file.
Multiple-to-multiple mode: an input file with several structures will be transformed into another multiple structure file. ![]() Multiple mode.
Multiple mode.
![]() The split mode (multiple-to-many) is available (Save window} only if a multiple file was previously inputted in multiple mode: instead of writing one new file with several structures, each one will automatically be written into a new file (the opposite of merge mode).
The split mode (multiple-to-many) is available (Save window} only if a multiple file was previously inputted in multiple mode: instead of writing one new file with several structures, each one will automatically be written into a new file (the opposite of merge mode).
| File type: | Structural levels:* Molecule/ Chain/ Residue |
Selection: | Multiple input | Multiple write, split and merge modes | Browse mode |
||
| in normal input | in multiple input | level | |||||
| Sybyl mol2 | M/C/R | all or selected M | no | 1 | yes | yes | yes |
| Insight | M/C/R | all or selected M | no | 1 | yes | yes | yes |
| MacroModel, Maestro | M/C/R | all or selected M | no | 1 | yes | yes | yes |
| PDB | M/C/R | all or selected M/C | yes | 2 | yes | yes | yes |
| HyperChem | M/R | all or selected M | no | 1 | yes | yes | yes |
| PCMODEL | M | one selected | no | 1 | yes | yes | yes |
| Xmol | M | all or selected | no | 1 | yes | yes | yes |
| MXYZ | M | all or selected | no | 1 | yes | yes | yes |
| MDL SD | M | one selected | no | 1 | yes | yes | yes |
| Cambridge MODEL | M | one selected | no | 1 | yes | no | yes |
| Cambridge FDAT and MODEL |
M | one selected | no | 1 | yes | no | yes |
| Multiple CIF (CIF/MIF) | M | one selected | no | 1 | yes | no | yes |
| Beilstein's Rosdal | M | one selected | no | 1 | yes | no | yes |
| Free format Z-matrix and Cartesian |
M | one selected | no | 1 | no | no | no |
Connectivity and bond types
Some molecules files, like most crystallographic files, Z-matrices or most of the PDB files, do not contain connectivity information at all. In this case Mol2mol will generate the connectivity matrix (bonds) using the standard covalent atom radii and valences. In the case of unusual valences, distances or bad geometry there may be mistakes in the created molecule. You can correct these in the Graphics window.
In addition, in most formats "natural" bond orders are used: 1, 2 and 3. In high-end modelling programs, having a built-in force-field or another advanced modules, other bond types (aromatic, amide, complex etc) are also used, designated usually with numbers greater than 3. Drawing programs may use a number of other types (bold, up/down wedged, hashed etc), cf. summary of stereo bond types.
Colouring of the different bonds in the graphics window (in high or true colour modes):
| 1-2-3 | white | oooo |
| amide | cyan | nnnn |
| aromatic | water green | nnnn |
| complex | light magenta | nnnn |
| ionic | yellow | nnnn |
| weak | orange | nnnn |
| dummy | pink | nnnn |
| no connection | pink | nnnn |
| unknown | pink | nnnn |
Conversion of special bonds
 Input structures containing complex bonds (Cache, MacroModel, PCModel), ionic or weak bonds (Cache) can be converted from group-2 to group-1 files easily. Alternatively, you could convert the complex bonds into single bonds or remove them altogether. With upward conversions, if the target program does not support these bond types and you choose the conversion to single bonds, the program may present some warning messages about unusual atoms, but this should not affect the target structure. If you choose to remove the complex bonds, the target modelling program may detect unconnected lone atoms and display error messages. The Cu-complex in the sample file, PCMOD.PCM, (the fourth structure) contains complex bonds. Try experimenting with it.
Input structures containing complex bonds (Cache, MacroModel, PCModel), ionic or weak bonds (Cache) can be converted from group-2 to group-1 files easily. Alternatively, you could convert the complex bonds into single bonds or remove them altogether. With upward conversions, if the target program does not support these bond types and you choose the conversion to single bonds, the program may present some warning messages about unusual atoms, but this should not affect the target structure. If you choose to remove the complex bonds, the target modelling program may detect unconnected lone atoms and display error messages. The Cu-complex in the sample file, PCMOD.PCM, (the fourth structure) contains complex bonds. Try experimenting with it.
Summary of stereo bond types
The following table illustrates the special stereo bond types in ISIS mol, PLT, ChemDraw ct, ChemDraft and Rosdal files. Their interconversion from one file format into another is possible, if an appropriate matching type can be found.

|

|
Summary of supported program molfiles
Atom Atom Bond Dummy Lone Default Multiple Chains, types symbols types atoms pairs extension Structures Residues ALCHEMY x 123mad x x MOL SYBYL # # 123madn x x SY2,MOL2 x x ISIS mol x 123(a)s x x MOL ISIS sdf x 123(a)s x x SDF x CSSR x 1 x CSS DTMM x 1 x MOL MOBY x (123a?) x x MOL Insight # # 123a x CAR x x MacroModel x 123cd x x OUT x x Maestro x x 123cd x x MAE x x Cache # # 123ciw x CSF HyperChem x 123a x x HIN, HCS x x PCModel x 123c x x PCM x Z matrix x ? Z Cartesian x ? ? CAR CACAO x IN PDB # # 1iw*(?) x PDB, ENT x x ROSDAL x 123 STR x ChemDraft x 123s x DAT MOPLO x 1 XYZ ChemWindow mol x 123a MOL Xmol x XYZ x MXYZ x 123 XYZ x CIF x 1 CIF CIF/MIF x 123a CMF x FDAT x 1 DAT x MODEL x 123a MDL SHELX x x SHX, INP PLUTO x 1 ? INP PLT x 123s x x PLT WIMP x 123 x x FTR SCHAKAL x 1 DAT ChemDraw CT x 123s x x CT POV-ray x any x x POV UltraMol (MolPic) x 123a MP3 Molecules 3D x 123aw x M3D a aromatic bonds m amide bonds n no connection c complex bonds d dummy bonds i ionic bond w weak bond s several stereo bond types are used (wedged, hashed etc) * for standard residues connectivity is not necessary ? depending on the version # both atom types and symbols are usedIn a few programs atom numbers are used instead of atomic symbols (6 for C, 8 for O, etc)
Summary of coordinates, rotation and translation methods
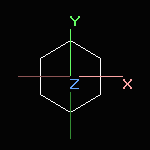
|
Screen rotation |
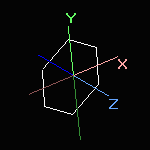
|
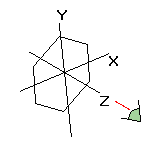
|
→ → → |
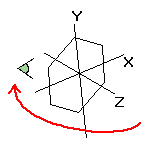
|
|
|
Simple rotation or translation with the mouse or by using the rotation sidebar changes the screen coordinates only.
 Simple translation is never applied to the world coordinates. |
If you want to apply the rotation to the original coordinates, do one of the followings:
a) From the Preferences 1 option dialog select the Write rotated coordinates radio button. Only the outputted coordinates are changed, not within the program. b) Select the Apply rotation to world option from upper button bar in the graphics window. The original coordinates are changed within the program. |

|
![]() Advanced, special rotation and translation modes:
Advanced, special rotation and translation modes:
![]() Centering methods from the upper button bar:
Centering methods from the upper button bar:

|
|
In the case avarage organic molecules the results of 3. and 5. are nearly the same. However, if the molecule contains one or more heavy atoms (Br, I etc) the difference can be profound. Experiment with the sample molecule TRIANONE.MOL.
Methods 3. and 4. are available from the main menu Utilities | Center molecule too.
![]() Centering and rotation methods from the rotation side bar:
Centering and rotation methods from the rotation side bar:
If you press the More button of the above rotation toolbar, new options appear:
|
|
|

![]() These 5 options always change the world coordinates!
These 5 options always change the world coordinates!
![]() The current state of the coordinate systems is reflected by the
The current state of the coordinate systems is reflected by the ![]() indicators in the upper status bar of the main window. Red lights reflect that the word and/or screen coordinates have been changed.
indicators in the upper status bar of the main window. Red lights reflect that the word and/or screen coordinates have been changed.
![]() By combining these methods, many transformations can be applied on the molecule. For example, if you want to rotate a given bond parallel to the X axis:
By combining these methods, many transformations can be applied on the molecule. For example, if you want to rotate a given bond parallel to the X axis:
![]() Do not manipulate world coordinates, if it is important to retain them unchanged
Do not manipulate world coordinates, if it is important to retain them unchanged


